医疗器械临床试验中的事件、SUDE和SUE——这在实践中意味着什么?
在即将进行的医疗器械临床试验或临床试验中,必须系统地记录事故或不良和严重不良事件。最大的挑战不是实际的报告,而是关于应记录和跟踪哪些事件以及在何种程度上采取后续行动的决定。这很重要,因为相关定义的性质和范围以及对监管要求的遵守既确保了所有研究参与者的安全,又是一个重要的成本因素。
本文旨在阐明基本术语和背景,并作为处理不良事件和事件的实用决策工具。
1) 术语及其关系和重叠之处的解释
MDR、MPDG 和 ISO14155 对产品缺陷、事故、(严重)不良事件和(严重)不良医疗器械影响的定义进行了规定。
一个 不良事件 (欧盟;英语:不良事件,AE)是根据第 2 条第 57 款规定的 MDR 在临床试验中发生的任何不良医疗事件,无论是否由研究设备引起。例如,这些症状包括研究参与者出现新的或恶化的症状、意外疾病、受伤或异常的实验室发现。
如果赞助方要求,只要研究参与者报告了特定症状或研究小组以其他方式发现了新疾病或新的辅助疗法,就可以根据测试计划记录活动并进行随访。
一个 严重不良事件 (SUE;英语:严重不良事件,SAE)是指根据MDR第 2 条第 58 款具有以下后果之一的不良事件:
1) 死亡,
2)受试者的健康状况严重恶化,这反过来又产生了以下后果之一:
· 危及生命的疾病或伤害,
· 永久性身体损伤或身体机能永久受损,
· 住院治疗或延长患者的住院治疗,
· 预防危及生命的疾病或伤害或永久性身体损伤或永久身体机能受损的医疗或外科干预,
· 慢性病,或
· 胎儿危害、胎儿死亡、先天性身体或精神障碍或出生缺陷。
一个 产品短缺 (下午; 英语:设备缺陷,DD) 对应于大声 MDR 第 2 条第 59 款 识别性、质量、耐久性、可靠性、安全性不足,以及市场上已上市产品的特性或性能出现故障或退化,包括制造商提供的信息不足或不利的副作用。根据第 11 节 (MPDG),只能在不危及患者、用户和第三方的安全和健康的情况下操作和使用医疗器械。
医疗器械的不良影响(嗯; 英语:不良设备效应,ADE 要么 事件) 发生在与使用研究设备相关的不良事件时。这些事件尤其包括因使用说明不足或不准确、设置、植入、安装或功能不正确,以及测试产品故障并导致不良事件而导致的事件,或 本可以领先 (MPDG)。
一个 严重的医疗器械不良影响(英语:严重不良设备效应、SADE 或严重事件,SI) 是严重的不良事件, 这与测试产品有因果关系,会导致典型的后果(例如死亡、危及生命的疾病、永久或严重残疾、住院或延长住院时间)或 本来可以导致 (MPDG)。
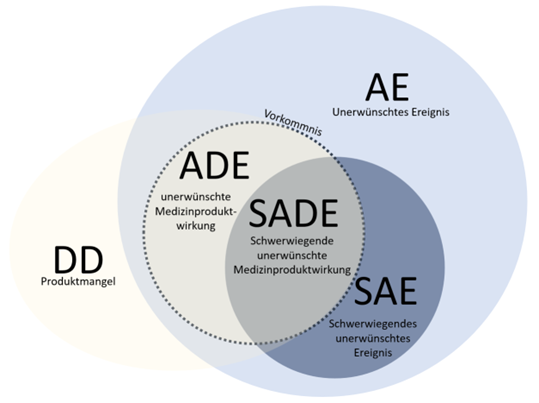
图 1:事件和事件的关联和重叠之处。
不良事件(AE)包括临床试验中的所有医学不良事件。根据严重程度,区分 AE(不严重)和 SAE(严重)。AE 与产品缺陷 (DD) 之间的因果关系被称为事件,即使仅是潜在损害(MPDG § 4)。根据严重程度,还会区分医疗器械不良反应(ADE)和严重不良医疗器械影响(SADE)。
2) 严重程度和因果关系的分类
因果关系是根据 MDCG 2020-10/1 修订版 1 标准进行评估的:
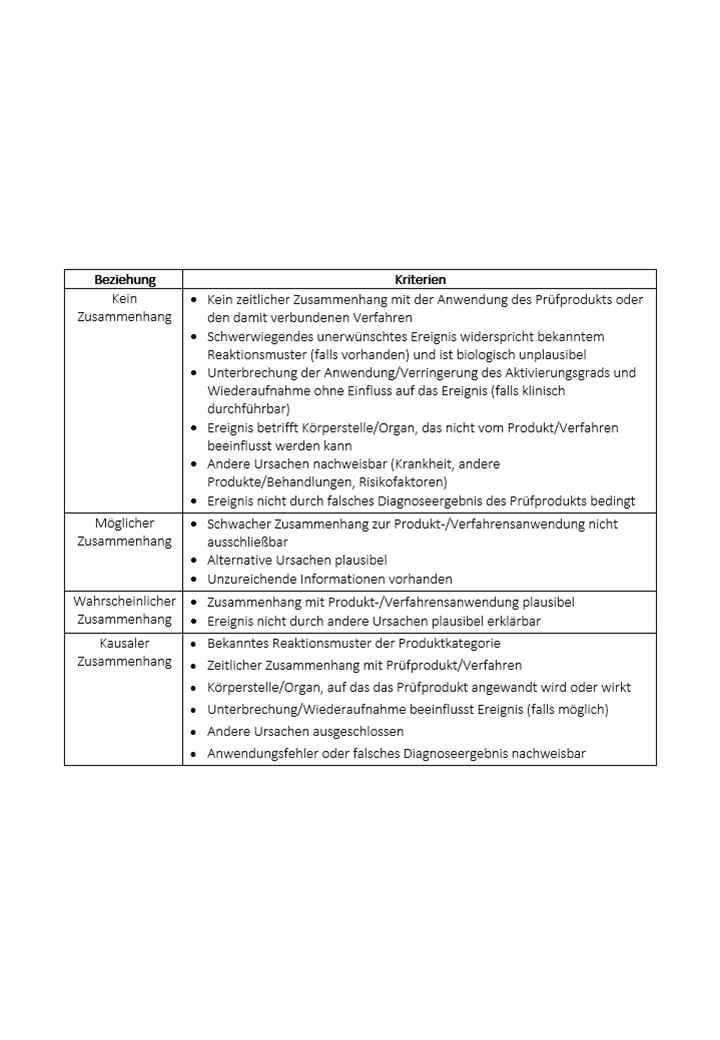
根据产品/工艺的性质和严重的不良事件,可能不必同时满足上述所有标准即可建立联系。
为了标准化 评估将是事件的严重程度 和不良事件可以按5分制进行分类。例如,该分类与因果关系无关,可以根据CTCAE、ICD 10、ICD 11或Medra进行相应的分类。这有助于对临床相关性和报告要求进行分类。
· 1 年级 (轻度):无症状或轻微症状;仅限临床或诊断观察;无或轻微损伤;未指干预。
· 二年级 (中度):建议进行最低限度、局部或非侵入性的干预;限制日常生活中适合年龄的器械活动。
· 三年级 (严重):医学意义重大,但不会立即危及生命;需要住院或延长住院治疗;残疾;为自我保健而限制日常生活活动。
· 四年级 (危及生命):对生命构成严重危险;表示需要紧急干预。
· 五年级 (死亡):事件造成的死亡。
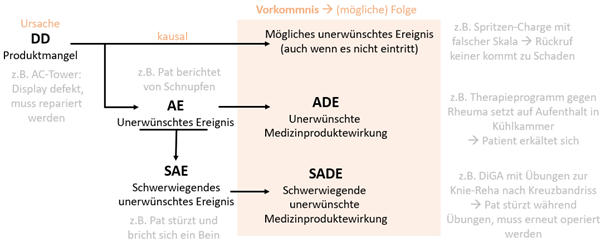
图 2:事件和不良事件的关系和重叠之处。
可能导致患者伤害的产品缺陷 (DD) 被称为事件(产品缺陷 = 原因,患者潜在损害/损害 = 后果)。如果 DD 和 AE 之间存在因果关系,则称为医疗器械不良效应 (ADE)。如果 DD 导致或可能如此,则 SAE 是严重的不良医疗器械效应 (SADE)。
评分:
· 研究人员对事件进行医学评估(严重程度、因果关系、预期/意外)并对其进行分类。
· 研究小组(研究人员、协调员)根据测试计划提供文件支持,记录在eCRF和内部表格中。
· 发起人检查分类,可以提问,并负责在适当时候向当局/道德部门报告。
-> 医疗 初始分类 由审计师提出,监管部门 报告和最终分类的责任 与赞助商或制造商/赞助商代表共处。
3) 文件和报告要求
临床试验中必须系统地记录和评估所有事件和不良事件,以监测参与者的安全性并评估研究设备的益处风险比。临床试验中的事件、严重不良事件 (SAE) 和产品缺陷 (DD) 根据欧洲法规 2017/745 (MDR) 以及《医疗器械法实施法》(MPDG) 进行记录和报告。
消息路径
· 审计师/首席调查员立即向发起人报告事件、SAE或DD。
· 发起人根据MDR/MPDG在适当时候(2或7天)向联邦主管当局报告应报告的事件、SAE和DD。
· 在充分使用EUDAMED之前,德国的传输以电子方式通过电子邮件发送到bFarm指定的职能邮箱(例如 mpsae@bfarm.de) 使用指定的表单。
· 对于已经上市的产品(警惕,研究之外保持警惕),制造商根据MDR第87条,通过预期的电子系统向主管当局报告该领域的严重事故和安全纠正措施,该系统通常由制造商所在成员国的当局进行协调。
事故/SAES报告后的典型纠正措施包括:
- 更新患者信息和同意文本(补充警告,经调整的风险描述)。
- 调整审计计划(例如额外的控制措施、更密切的后续行动、更改纳入/排除标准)。
- 对调查人员和用户进行培训或再培训,使其了解如何正确处理他们并最大限度地降低风险。
- 个人或所有测试对象中断或停止使用测试设备。
- 暂时停止招聘或暂停研究。
- 产品或配件的技术或组织变更(例如其他安全信息、更改的标签或操作说明)。
- 现场安全纠正措施 (FSCA),例如召回特定批次、更换组件、软件更新或向用户发出安全通知。
您想在医疗器械的临床研究/测试中保持安全和适应风险的警惕吗? 联系我们 请随时联系我们进行初步咨询!
MEDIACC
展示您的产品的医疗益处
凭借我们多年的经验和专业知识,我们提供有效的解决方案,以展示您的产品的医疗益处。
从临床前和临床研究的构思到实施,我们为您提供量身定制的服务。
了解 MEDIACC 如何帮助您获得产品退款。