Incidents, SADE and SAE in clinical investigations of medical devices — what does that mean in practice?
In upcoming clinical studies or clinical investigations of medical devices, incidents or adverse and serious adverse events (SAE) must be systematically documented under the European MDR. It is not the actual report that is the biggest challenge, but the decision as to which events should be documented and followed up and to what extent. This is important because the nature and scope of the relevant definitions and compliance with regulatory requirements both ensure the safety of all study participants and represent a significant cost factor.
This article is intended to clarify the underlying terminology and context and serve as a practical decision-making tool for dealing with adverse events (AE) and incidents.
1) Explanations of terms, their relationships and overlaps
Definitions for product deficiency, incidents, (serious) adverse events and (serious) adverse medical device effects are regulated in European Regulation 2017/745 (MDR) in conjunction with the Medical Device Law Implementation Act (MPDG, for Germany) and ISO14155.
An adverse event (AE) in accordance with MDR Article 2 paragraph 57 is any adverse medical event that occurs as part of a clinical investigation, whether or not it was caused by the investigational device. These include, for example, new or worsening symptoms, unexpected illnesses, injuries or unusual laboratory findings among study participants.
If requested by the sponsor, an event can be documented and followed up in accordance with the investigational plan as soon as the study participants (subjects) report a specific symptom or the investigational team otherwise detects a new disease or a new adjunctive therapy.
A serious adverse event (SAE) means an adverse event which had one of the following consequences in accordance with Article 2 paragraph 58 MDR:
1) death,
2) severe deterioration in the health status of the subject, which in turn had one of the following consequences:
· life-threatening illness or injury,
· permanent physical damage or permanent impairment of a bodily function,
· inpatient treatment or extension of the patient's inpatient treatment,
· medical or surgical intervention to prevent a life-threatening illness or injury or permanent physical damage or permanent impairment of bodily function,
· chronic illness, OR
· Fetal hazard, fetal death, or congenital physical or mental impairment or birth defects.
A Device Deficiency (DD) according to Article 2, paragraph 59 MDR means an insufficiency in identification, quality, durability, reliability, safety, and a malfunction or degradation of the characteristics or performance of a product already made available on the market, including inadequate information provided by the manufacturer or an adverse device effect. According to § 11 MPDG, medical devices may only be operated and used in such a way that the safety and health of patients, users and third parties are not endangered.
Adverse device effects (Adverse Device Effect, ADE or Incident) occur when there is an adverse event associated with the use of an investigational device. These include, in particular, events that result from inadequate or inaccurate instructions for use, incorrect set-up, implantation, installation or function, as well as from malfunctions of the test product and have led to adverse events, or could have led (MPDG) to AE.
A serious adverse device effect (SADE or Serious Incident, SI) is a serious adverse event, which is causally related to the test product and leads to the typical consequences (e.g. death, life-threatening condition, permanent or significant disability, hospitalization or extension of a hospital stay) or could have led (MPDG) to a SAE.
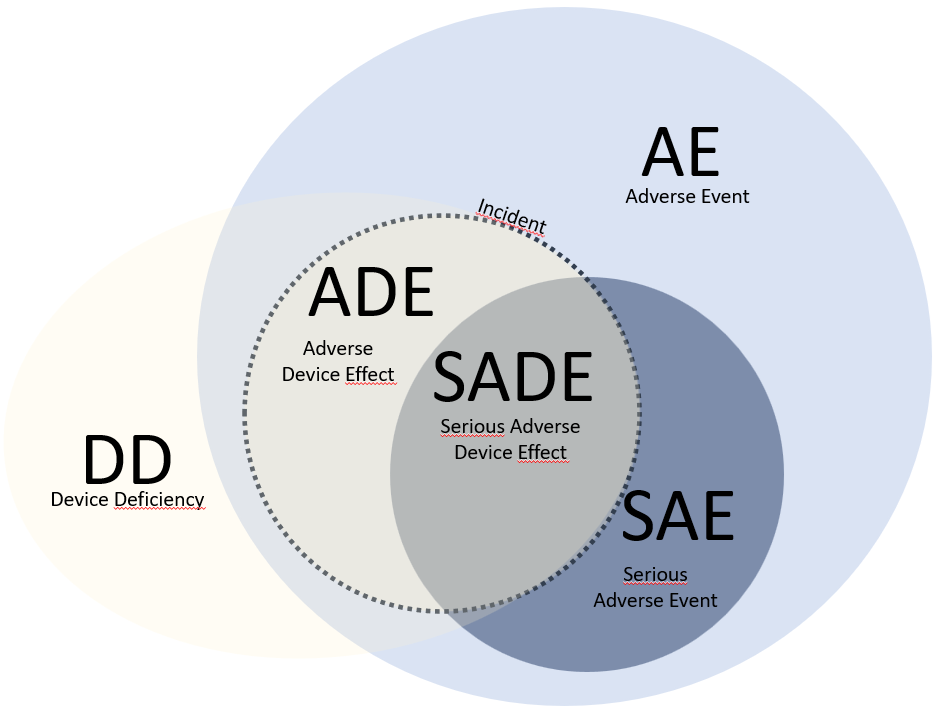
Figure 1: Connections and overlaps of incidents and events.
Adverse events (AE) include all medically adverse events in clinical trials. Depending on the severity, a distinction is made between AE (not severe) and SAE (severe). A causal relationship between an AE and a product defect (DD) is referred to as an incident, even in the case of only potential damage (MPDG § 4). Depending on the degree of severity, a distinction is also made between adverse device effects (ADE) and serious adverse device effects (SADE).
2) Classification of severity and causality
Causality is assessed according to MDCG 2020-10/1 Rev. 1 criteria:
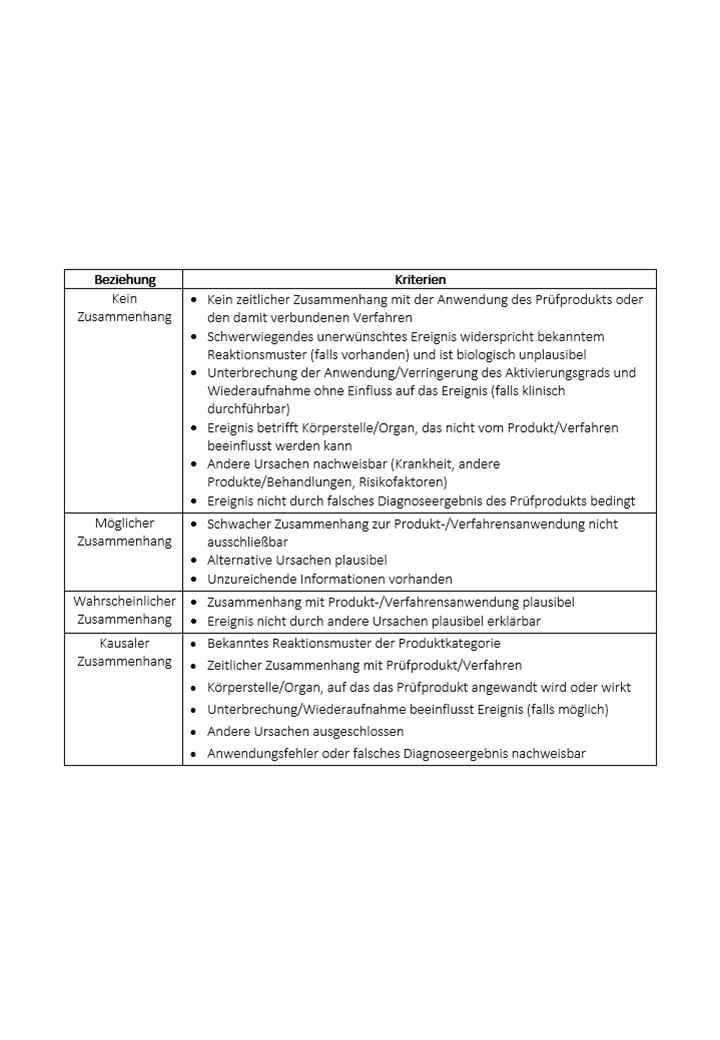
Depending on the nature of the product/process and the serious adverse event, it may not be necessary to meet all of the criteria listed above at the same time to establish a causality.
To standardise the assessment of the severity, incidents and adverse events are classified using a 5-point scale. The classification is independent of causality and can be categorised according to i.e. CTCAE, ICD 10, ICD 11 or MedDRA. This helps to classify clinical relevance and reporting requirements.
· Grade 1 (Mild): asymptomatic or mild symptoms; clinical or diagnostic observations only; no or minimal impairment; intervention not indicated.
· Grade 2 (Moderate): minimal, local or non-invasive intervention indicated; restriction of age-appropriate activities in daily life.
· Grade 3 (Serious): medically significant but not immediately life-threatening; inpatient admission or extension of inpatient treatment indicated; disabling; restriction of activities of daily living for self-care.
· Grade 4 (Life-threatening): acute danger to life; urgent intervention indicated.
· Grade 5 (death): Death as a result of the event.
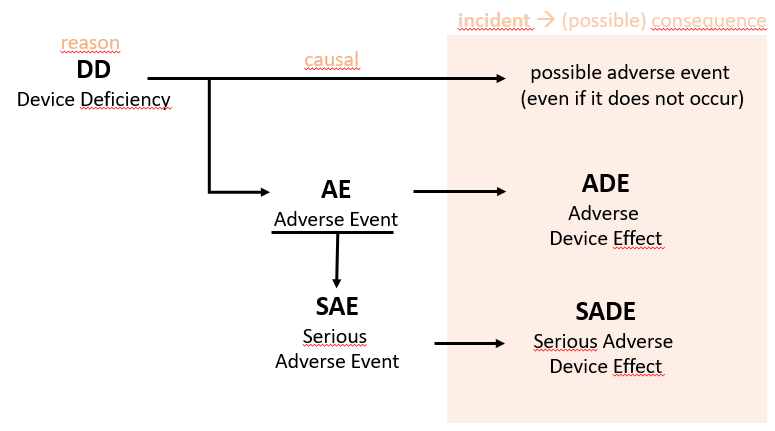
Figure 2: Relationships and overlaps of incidents and adverse events.
A device defiency (DD) that can potentially lead to patient harm is referred to as an incident (device deficiency = cause, patient damage/damage potential = consequence). If there is a causal relationship between DD and AE, this is referred to as an adverse device effect (ADE). If DD results in or could result in an SAE, this is a serious adverse medical device effect (SADE).
Process os Rating:
· Investigator evaluates the event medically (severity, causality, expected/unexpected) and classifies it.
· The study team (study staff, coordinators) provides support with documentation, recording in eCRF and internal forms in accordance with the investigational plan.
· The sponsor checks the classification, can ask questions and is responsible for reporting to authorities/ethics in due time.
-> Medical initial classification by a qualified medical doctor,
-> Regulatory responsibility for reporting and final classification with the sponsor or manufacturer/sponsor representative.
3) Documentation and reporting requirements
All incidents and adverse events must be systematically documented and evaluated in clinical trials in order to monitor the safety of participants and to assess the benefit‑risk ratio of the investigational device. Incidents, serious adverse events (SAEs) and product deficiencies (DD) in clinical trials are documented and reported in accordance with European Regulation 2017/745 (MDR) in conjunction with the Medical Device Law Implementation Act (MPDG).
Message path
· Auditor/Principal Investigator immediately reports an incident, SAE, or DD to the sponsor.
· The sponsor reports the reportable incidents, SAEs and DDs in due time (2 or 7 days) to the competent higher federal authority in accordance with MDR/MPDG.
· Until EUDAMED is fully usable, reporting in Germany is done electronically by email to the functional mailbox specified by the BfArM (e.g. mpsae@bfarm.de) using the specified forms.
· For products already on the market (vigilance, outside of studies), manufacturers report serious incidents and safety corrective measures in the field in accordance with Article 87 MDR to the competent authorities via the intended electronic system, usually coordinated by the authority of the Member State in which the manufacturer is based.
Typical corrective measures following an incident/SAES report include:
- Updating patient information and consent texts (supplemented warnings, adapted risk description).
- Adjustment of the audit plan (e.g. additional controls, closer follow-up, changed inclusion/exclusion criteria).
- Training or retraining of investigators and users on how to handle them correctly and minimize risks.
- Interruption or discontinuation of use of the investigational device by individual or all subjects.
- Stop recruiting temporarily or pause the clinical investigation.
- Technical or organisational changes to the product or accessories (e.g. additional safety information, changed labeling or operating instructions).
- Field safety corrective action (FSCA), e.g. recall of specific batches, replacement of components, software update, or security notice to users.
Would you like safe and risk-adapted vigilance for the clinical study/testing of your medical device? Feel free to contact us for an initial consultation!
MEDIACC
Show the medical benefits of your product
With our many years of experience and expertise, we offer effective solutions to demonstrate the medical benefits of your product.
From the conception to the execution of preclinical and clinical investigations, we support you with customized services.
Find out how MEDIACC can help you achieve reimbursability for your products.